
Druck-Version
Diskussionsbeitrag
Gen für das Großhirn: Die rasche Evolution des HAR1F-Gens
Stellt die HAR1-Region die Evolution des Menschen infrage?
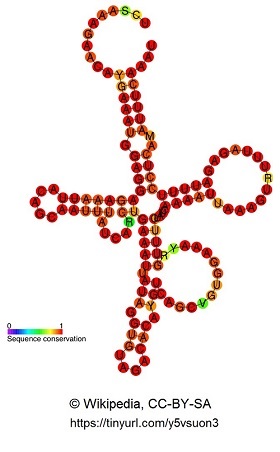
Die HAR1-Region ist eine DNA-Sequenz im menschlichen Genom, die regulatorische Aufgaben bei der Hirnentwicklung übernimmt. Sie ist Teil von zwei überlappenden Genen, von denen eines, das HAR1F-Gen, in der frühen Embryonalentwicklung des Menschen in den sogenannten Cajal-Retzius-Nervenzellen des Neocortexes aktiv ist. Dort exprimiert es ein RNA-Molekül, das durch Faltung eine bestimmte 3D-Struktur bildet.
Vermutlich aktiviert das Molekül wie ein Schlüssel bestimmte Gene und spielt so eine entscheidenden Rolle bei der Bildung der geschichteten Struktur der Großhirnrinde des Menschen. Die betreffende Region ist jedoch uralt, sie existierte bereits vor 3-400 Mio. Jahren und ist fast während der gesamten Vertebraten-Evolution hochkonserviert geblieben. Erst innerhalb der letzten Millionen Jahre kam in unserer Vorfahren-Linie Bewegung in die Sache, mit einschneidenden Konsequenzen.
Titelbild: Sekundärstruktur und Konservierungsgrad der einzelnen Nukleotide von HAR1A. Bildquelle: http://en.wikipedia.org/wiki/User:Ppgardne, HAR1F RF00635 rna secondary structure, CC BY-SA 3.0.
{kind=link}
Rasche Evolution
Das HAR1F-Transkript umfasst 118 Basenpaare. Der Vergleich der HAR1F-Varianten bei Schimpanse und Huhn offenbart einen Unterschied in nur zwei Positionen, was bedeutet, dass sich das Gen in den letzten 300 Mio. Jahren so gut wie nicht veränderte. Zwischen Schimpanse und Mensch hingegen sind die Unterschiede dramatisch: 18 DNA-Bausteine wurden ausgetauscht. Während der Menschwerdung fand also eine massiv beschleunigte Evolution statt (daher der Name: HAR = human accelerated regions). Offenbar beeinflusste die veränderte Genregulation die Hirnentwicklung signifikant und half unseren Hominini-Vorfahren bei der Menschwerdung. Dies ist womöglich einer der genetischen Gründe, der uns Menschen in geistiger Hinsicht erst zu dem macht, was wir sind.
BENIAMINOV et al (2008) untersuchten die HAR1F-Raumstruktur des Menschen und des Schimpansen und stellten fest, dass sich die RNA-Moleküle in zwei verschiedene 3D-Strukturen falten können, nämlich in eine eher gestreckte und eine kleeblatt-förmige. Es scheint, dass die menschliche Form vornehmlich in einer "Kleeblatt-Faltung" vorliegt und die des Schimpansen vornehmlich in gestreckter Faltung. Die 18 Mutationen sorgen unter anderem dafür, dass sich zwei benachbarte Schleifen um zwei Basen verschieben – und dabei die Kleeblatt-Faltung stabilisieren. Dies könnte dazu führen, dass eine Genkaskade anders aktiviert wird, sodass die Hirnentwicklung beim Menschen in einzigartiger Weise anders verläuft als bei den übrigen Primaten. Die Details hierzu sind allerdings noch nicht geklärt.
Was aber ist die Ursache für die beschleunigte Evolution in der HAR-Region? POLLARD et al. (2006) weisen darauf hin, dass eine Antwort bislang ausstehe. Dies führt dazu, dass Evolutionsgegner Berechnungen anstellen, um die evolutionäre Entwicklung des Menschen in Frage zu stellen (BORGER 2020). Wie sieht dieses Argument aus?
Das Unwahrscheinlichkeits-Szenario
BORGER wählt einen populationsgenetischen Ansatz, um seinem Argument eine mathematische Präzision zu verleihen. Als Ausgangspunkt dient die Erkenntnis, dass der letzte gemeinsame Vorfahr von Mensch und Schimpanse vor etwa 6-7 Millionen Jahren lebte. Die Evolution hatte also maximal 7 Millionen Jahre Zeit, um die menschliche Variante des HAR1F-Gens entstehen zu lassen. So weit, so gut. Des Weiteren geht der Autor davon aus, dass die effektive Populationsgröße während dieser 7 Millionen Jahre durchschnittlich bei etwa 10.000 Individuen lag. Auch das erscheint korrekt. Wie groß sei nun die Chance, eine Mutation "an der richtigen Stelle" im HAR1F-Gen zu erhalten? BORGERs Antwort:
"Pro Individuum besteht die Chance von 1 zu 30 Millionen, dass eine der oben erwähnten 100 Mutationen pro Generation an die richtige Stelle fällt (100/3 Milliarden = 1/30 Millionen). Die Chance, dass dies einmal bei einer Population von 10.000 Menschen (oder ihren mutmaßlichen Vorfahren) geschieht, liegt demnach bei 1/3000. Mit anderen Worten, alle 3000 Generationen wird es durchschnittlich einmal passieren. Wenn wir durchschnittlich 10 Jahre für eine Generation rechnen, dann dauert es 30.000 Jahre, um einmal einen Treffer zu haben. Aber ist es der richtige Treffer? Es muss auch der richtige DNA-Buchstabe sein (die DNA hat vier verschiedene Nukleotide).1 In zwei von drei Fällen ist es der falsche Buchstabe. Bevor wir also den ersten richtigen Treffer landen, sind 30.000–90.000 Jahre vergangen!"
Im Weiteren rechnet BORGER vor, wie gering die Überlebens- bzw. Fixierungswahrscheinlichkeit einer solchen Mutation nach populationsgenetischen Gesichtspunkten sei:
"Nach Ansicht der Populationsgenetiker hat jede neutrale Mutation nur die minimale Chance ..., dass diese Mutation nicht wieder verloren geht! Nachdem eine Mutation endlich an der richtigen Stelle aufgetreten ist, ist die Chance, dass sie tatsächlich in der Population erhalten bleibt, also minimal (1/20.000 bei einer Populationsgröße von 10.000 Individuen). Hier braucht man daher die natürliche Auslese. Wir müssen also davon ausgehen, dass die Punktmutation (Austauschs eines Nukleotids) im HAR1F-Gen des Vorfahren (das bei den oben erwähnten Affen monomorph ist, also bei allen Individuen identisch und nicht mutiert) einen selektiven Wert hat. Das ist durchaus möglich. Geben wir dieser Mutation einen Selektionsvorteil von 0,5% gegenüber dem Gen des Vorfahren, ist das 'leicht vorteilhaft', aber dennoch sehr großzügig für eine Punktmutation in einem stabilen Gen (eher erwartet man einen Nachteil). Die Mutation hat nach populationsgenetischen Berechnungen nun eine Chance von 1%, sich in der Bevölkerung zu etablieren.
Ein Treffer muss also durchschnittlich 100 Mal erfolgen, um sich dauerhaft einmal in der Population anzusiedeln. Der erste richtige Treffer tritt erst nach 30.000–90.000 Jahren ein, aber in der Population muss dies durchschnittlich 100 Mal geschehen! 30.000–90.000 x 100 ergibt 3–9 Millionen! Es dauert daher 3–9 Millionen Jahre, um eine Mutation mit 0,5% Selektionsvorteil durch Selektion dauerhaft im HAR1F-Gen zu erhalten! Es werden aber noch weitere 17 Mutationen im HAR1F-Gen benötigt."
Daraus folgert der Autor, "zufällige Mutationen, Selektion und Gendrift" könnten die Herkunft des menschlichen HAR1F-Gens "nicht erklären"; der Zeitraum von 6-7 Mio. Jahren sei dafür zu kurz. Entsprechendes gälte für die 2700 weiteren HAR im menschlichen Genom. Eine "schrittweise Evolution", so das apodiktisch zugespitzte Resümee, sei "sicherlich nicht die richtige Erklärung unserer Existenz!".
Die Kritik
Der Populationsgenetiker Professor Martin HASSELMANN von der Universität Hohenheim äußerte sich für die AG Evolutionsbiologie zur Argumentation der (wie er sagt) "Genesisnet-Leute" (E-Mail vom 29.06. und vom 11.07.2020). Aus seiner Sicht ist BORGERs Behauptung, man könne auf "einfache Weise berechnen, ob das menschliche HAR1F-Gen auf darwinistische Weise – durch Mutation, Selektion und Gendrift – entstehen konnte oder nicht", grundlegend verfehlt. (Zur detaillierten Kritik siehe BEYER 2020).
Zum einen bediene sich die Kalkulation vereinfachter Parameter, wie etwa einer Populationsgröße, eines Selektionsvorteils und einer Fixierungschance, die nicht präzise anzuwenden sind, da sie schlicht unbekannt sind. Zum anderen sei "rapid-evolution"-Prozessen gerade nicht mit populationsgenetischen Standardansätzen beizukommen!
Die Kritikpunkte im Einzelnen:
1. Aus dem (vermeintlichen oder tatsächlichen) Fehlen einer "neo-darwinistischen" Erklärung folgt nicht, dass die schrittweise Evolution des Menschen zur Disposition steht. Abgesehen von einer noch ausstehenden Detail-Erklärung stützen so viele empirische Belege die Abstammung des Menschen aus dem Tierreich, dass keine vernünftigen Zweifel möglich sind (siehe: Evolutionsbeweis durch endogene Retroviren). Das sind zwei logisch voneinander unabhängige Sachverhalte, die es auseinanderzuhalten gilt!
2. BORGERs Annahme, die Evolution müsse warten, bis eine Mutation zufällig an "die (eine!) richtige Stelle" im Genom falle, hat einen entscheidenden Einfluss auf das Rechenergebnis. Sie ist insofern willkürlich, als sie im gegebenen Kontext überhaupt nur eine konkrete Mutation an einer ausgewählten Nukleotidstelle als "Treffer" gelten lässt! Damit setzt der Autor etwas voraus, was er nicht belegen kann. Vielmehr wissen wir von RNA-Molekülen, dass eine Vielzahl unterschiedlichster Sequenzen identische Strukturen hat (vgl. SCHUSTER 1995; 2006). Warum sollen, um beim Beispiel zu bleiben, nicht viele verschiedene Mutationen an den unterschiedlichsten Stellen im HAR1F-Gen die Kleeblatt-Struktur stabilisieren können? Überhaupt, warum sollte nur dieses RNA-Molekül die Vergrößerung der Gehirnkapazität ermöglichen? Verschiedenste Signalmoleküle und Gene nehmen Einfluss auf die Hirnentwicklung! Solange wir nicht den gesamten Möglichkeitsraum kennen, hat BORGERs "Berechnung" keinen Realitätsbezug.
3. Die Kalkulation lässt das parallele Auftreten von Mutationen und deren Zusammenführung durch meiotische Rekombination unberücksichtigt. Dieser Mechanismus ist gerade für HAR1 relevant, weil das Gen in der Nähe eines sogenannten Rekombinations-Hotspots liegt, wo Rekombinationen bis zu Tausend mal häufiger auftreten als im Rest des Genoms. Zudem kann in der unmittelbaren Umgebung der Hotspots ein bevorzugter Austausch und Einbau einzelner Nukleotide (biased gene conversion) zu einer hohen Mutationsrate führen. (Den letztgenannten Mechanismus erwähnt BORGER zwar, berücksichtigt ihn jedoch nicht in seinem stochastischen Ansatz.)
4. Die Populationsdynamik von Mutationen in Subpopulationen, die vielleicht auch noch durch "Flaschenhälse" gegangen sind und schwankender sexueller Selektion unterliegen, ist äußerst komplex. Gerade wenn Flaschenhals-Effekte auftreten, kann alles sehr schnell gehen. Eine Reduktion auf die üblichen populationsgenetischen Standardansätze ist daher nicht möglich.
5. Der Selektionskoeffizient von 0,5% für eine passende Mutation und der daraus abgeleitete Schluss, die Evolution müsse einen "Treffer" statistisch 100 Mal wiederholen, bevor er eine reelle Chance habe, sich in der Population auszubreiten, ist von BORGER willkürlich gewählt, um Evolution unwahrscheinlich zu rechnen. Bei einem Selektionskoeffizienten von 5% fällt das Ergebnis ganz anders aus. Gerade die Etablierung einer neuen, besonders vorteilhaften Funktion begünstigt die rasche Fixierung der betreffenden Mutationen.
6. Die Annahme, die vorteilhaften Mutationen müssten sich komplett in der Population ausbreiten (fixiert werden), ist ebenfalls nicht gerechtfertigt. Es genügt bereits, wenn die betreffenden Allele mit einer gewissen Häufigkeit in einigen Populationen vertreten sind, so dass Folgemutationen auftreten können oder eine Rekombination mit anderen, parallel aufgetretenen Mutationen stattfinden kann.
Wir könnten folgende (nicht weniger legitime) Gegenrechnung aufmachen: Jeder Mensch erbt statistisch 100 Mutationen von seinen Eltern. Dann wäre, eine Population von 10.000 Individuen vorausgesetzt, in jeder 10. Generation statistisch einmal das HAR1F-Gen von einer Mutation betroffen. Selbst wenn nur jede Tausendste (!) Mutation einen deutlichen Selektionsvorteil hätte (wahlweise: wenn 10% der Mutationen einen Selektionskoeffizienten von lediglich 0,5% aufwiesen), würde etwa alle 200.000 Jahre eine solche Mutation fixiert, und das von BORGER gelieferte Argument löst sich in Luft auf.
Fazit
Die Achillesferse in BORGERs Argument sind die willkürlich gewählten Voraussetzungen, die in ihrer Gesamtheit zutreffen müssten, damit seine populationsgenetische Abschätzung plausibel wäre. Zudem gibt er Modell-Parameter vor, die dabei "helfen", die Evolution unwahrscheinlich zu rechnen. In Wahrheit sind die Parameter und prähistorischen Bedingungen schlicht unbekannt und die Vorgänge so komplex, dass eine Reduktion auf die üblichen populationsgenetischen Standardansätze kaum möglich ist.
Wie bereits LEROY (1960, S. 9) in seinem Lehrbuch bemerkte, ist bei der Übertragung populationsgenetischer Modell-Rechnungen auf realhistorische Ereignisse größte Vorsicht geboten:
"In der Populationsgenetik stützen sich die numerischen Auswertungen von Versuchen und Erhebungen meist auf bestimmte, vorgängig festgelegte Hypothesen (zum Beispiel rein additive Genwirkung, keine Genotyp-Umwelt-Korrelation usw.). Das bedeutet eine gewisse Gefahr, einmal deswegen, weil das Ergebnis nur dann stimmt, wenn die richtige Hypothese zugrunde gelegt wurde (was in der betreffenden Untersuchung selbst gar nicht verifiziert werden kann). Dann aber auch deswegen, weil über den zumeist langwierigen Rechenoperationen die gemachten Voraussetzungen leicht vergessen werden, sodass dann die Ergebnisse unzulässig verallgemeinert werden."
Das ist das notorische Problem der Evolutionskritik; ihre Verfechter multiplizieren und potenzieren in einer Art und Weise, dass darüber völlig die Voraussetzungen vergessen werden, unter denen ihre Schlüsse berechtigt wären. Ihre Berechnungen zur Unwahrscheinlichkeit der Evolution lassen sich treffend mit der bekannten Phrase aus der Informatik "Garbage in, Garbage out" (kurz: GIGO) charakterisieren. Der WORT-UND-WISSEN-Debütant Peter BORGER ist da keine rühmliche Ausnahme.
Literatur
BENIAMINOV, A. et al. (2008) Distinctive structures between chimpanzee and human in a brain noncoding RNA. RNA 14, S. 1270–1275.
BEYER, A. (2020) Detailanmerkungen zu: "Das HAR1F-Gen stellt Evolution in Frage".
BORGER, P. (2020) Das HAR1F-Gen stellt Evolution in Frage. Letzter Zugr. a. 24.08.2020
LEROY, H. (1960) Statistische Methoden der Populationsgenetik. Verlag Birkhäuser, Basel.
POLLARD, K. S. et al. (2006) An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, S. 167–172.
SCHUSTER, P. (1995) How to search for RNA structures. Theoretical concepts in evolutionary biotechnology. Journal of Biotechnology 41, S. 239–257.
SCHUSTER, P. (2006) Prediction of RNA secondary structures. From theory to modelsand real molecules. Reports on Progress in Physics 69, S. 1419–1477.
Autoren: Martin Neukamm & Andreas Beyer
